








近日,家畜营养与调控科技创新团队利用16s rDNA测序技术研究猪肠道菌群的空间分布情况,揭示肠黏膜与肠腔、不同肠段微生物种属水平及功能组成的异质性,建立不同“生态位”上的菌群共生网络。
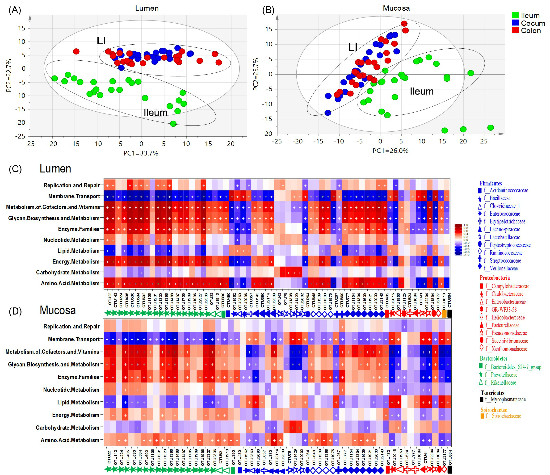
该研究描绘猪肠腔内菌群的空间分布,发现回肠与盲、结肠之间,肠腔与肠黏膜微生物之间的显著差异。通过SparCC共线性网络分析发现普雷沃式菌科、毛螺菌科以及韦荣氏球菌科在肠腔和肠黏膜微生物上都形成共现性微生物菌群,而来自于γ-变形菌门中机会性致病菌则仅在猪肠道黏膜上形成共现性微生物菌群。肠腔中肠杆菌科和以普雷沃式科为中心的菌群形成强的共排斥关系;冗余分析发现胆汁酸(37.1%)以及短链脂肪酸(41%)组成能解释肠腔微生物组成中51%的变化。部分核心菌群参与宿主必需氨基酸、能量、辅酶因子和维生素的代谢。结果表明猪肠道菌群空间分布的异质性以及共生模式是与其所在“生态位”的微环境紧密相关;核心微生物之间的互作对宿主肠道代谢和免疫发挥着重要作用。该成果可为靶向性的调控肠道菌群改善肠道健康提供重要的理论依据。
该研究成果以《生长猪肠腔微生物和肠黏膜微生物的空间分布异质性以及共现性网络分析》为题发表于著名期刊《微生物学前沿(Frontiers in Microbiology)》2018年1月第9卷。中国农业科学院北京畜牧兽医研究所为第一完成单位,博士生张莉为第一作者,解竞静副研究员和张宏福研究员为通讯作者。
原文链接:https://www.frontiersin.org/articles/10.3389/fmicb.2018.00048/full